AP Chemistry: Chemical Kinetics (Unit 5)
5.1 Reaction Rates
Chemical Kinetics is the study of the speed (rate) at which chemical reactions occur and the mechanisms by which they happen. It is distinct from thermodynamics; thermodynamics tells us if a reaction will happen (spontaneity), while kinetics tells us how fast it happens.
The Definition of Rate
The rate of a reaction is defined as the change in concentration of reactants or products per unit of time.
- Reactants disappear (negative change in concentration).
- Products appear (positive change in concentration).
For a generic reaction
The rate is expressed as:
Note: We divide by the stoichiometric coefficient to normalize the rate. If 1 mole of A produces 2 moles of C, C appears twice as fast as A disappears, but the "rate of reaction" is a single standardized value.
Factors Affecting Reaction Rate
- Concentration: Higher concentration $\rightarrow$ more frequent collisions $\rightarrow$ faster rate.
- Pressure (Gases): Increasing pressure (decreasing volume) increases concentration $\rightarrow$ faster rate.
- Surface Area (Solids): Greater surface area (powders vs. chunks) $\rightarrow$ more collision sites $\rightarrow$ faster rate.
- Temperature: Higher temperature $\rightarrow$ faster particle speed and higher energy $\rightarrow$ more frequent AND more effective collisions $\rightarrow$ significantly faster rate.
- Catalysts: Provide an alternate pathway with lower activation energy (speed up reaction).
5.2 & 5.3 Rate Laws
There are two ways to mathematically describe kinetics: Differential Rate Laws (Rate vs. Concentration) and Integrated Rate Laws (Concentration vs. Time).
The Differential Rate Law
This creates a relationship between the rate of the reaction and the concentration of reactants.
- $k$: The Rate Constant. It is temperature-dependent. It does not change with concentration.
- $m$ and $n$: The reaction orders. They represent the sensitivity of the rate to changes in concentration.
- These must be determined experimentally; they are NOT the coefficients from the balanced equation.
Method of Initial Rates (How to Determine Order)
To find the rate law, we look at how the initial rate changes when we vary the concentration of one reactant while holding others constant.
Common Orders:
- Zero Order ($x=0$): Rate is independent of [A]. Doubling [A] has no effect on rate.
- First Order ($x=1$): Rate is directly proportional to [A]. Doubling [A] doubles rate.
- Second Order ($x=2$): Rate is proportional to the square of [A]. Doubling [A] quadruples rate ($2^2=4$).
Worked Example
Given the data for :
| Exp | A | B | Initial Rate (M/s) |
|---|---|---|---|
| 1 | 0.1 | 0.1 | 0.01 |
| 2 | 0.1 | 0.2 | 0.02 |
| 3 | 0.2 | 0.1 | 0.04 |
- Find Order for B: Compare Exp 1 and 2. [A] is constant. [B] doubles ($0.1 \rightarrow 0.2$). Rate doubles ($0.01 \rightarrow 0.02$).
- $2^n = 2 \rightarrow n = 1$. First order with respect to B.
- Find Order for A: Compare Exp 1 and 3. [B] is constant. [A] doubles ($0.1 \rightarrow 0.2$). Rate quadruples ($0.01 \rightarrow 0.04$).
- $2^m = 4 \rightarrow m = 2$. Second order with respect to A.
- Write Law: Rate $= k[A]^2[B]^1$. Overall order $= 2+1 = 3$.
- Find $k$: Use Exp 1 data.
- $0.01 = k(0.1)^2(0.1)^1$
- $k = \frac{0.01}{0.001} = 10 \text{ M}^{-2}\text{s}^{-1}$
Units of the Rate Constant ($k$)
The units of $k$ change depending on the overall order of the reaction so that the rate always ends up as $M/s$.
| Overall Order | Units of $k$ | Shortcut Formula |
|---|---|---|
| 0 | $M \cdot s^{-1}$ | $M^{(1-\text{order})} \cdot s^{-1}$ |
| 1 | $s^{-1}$ | |
| 2 | $M^{-1}s^{-1}$ | |
| 3 | $M^{-2}s^{-1}$ |
5.3 Integrated Rate Laws
Integrated rate laws relate concentration and time. This allows you to calculate how much reactant is left after a certain period. The order of a reaction dictates which mathematical function produces a linear graph.
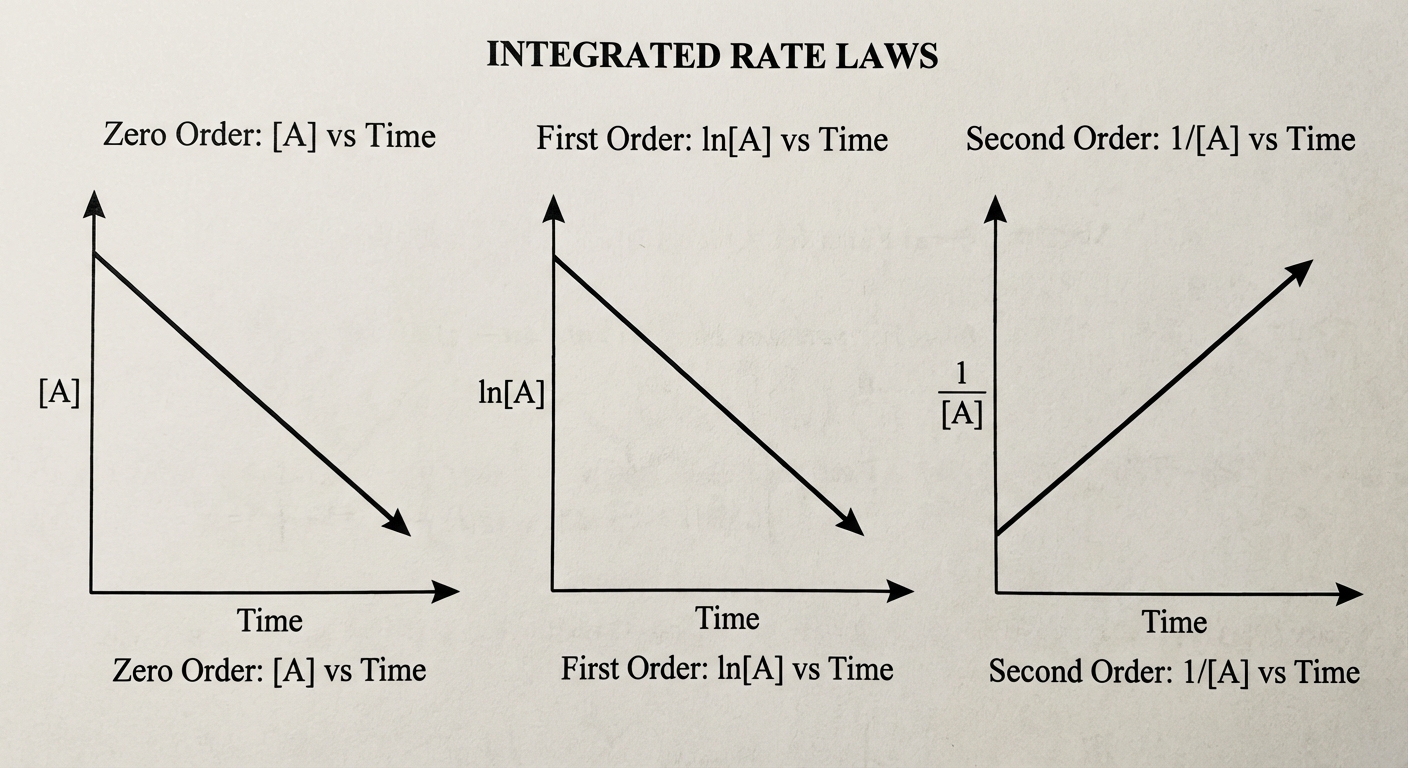
Summary Table (Memorize This)
| Feature | Zero Order | First Order | Second Order |
|---|---|---|---|
| Rate Law | Rate $= k$ | Rate $= k[A]$ | Rate $= k[A]^2$ |
| Integrated Law | $[A]t - [A]0 = -kt$ | $\ln[A]t - \ln[A]0 = -kt$ | $\frac{1}{[A]t} - \frac{1}{[A]0} = kt$ |
| Linear Graph | $[A]$ vs. Time | $\ln[A]$ vs. Time | $\frac{1}{[A]}$ vs. Time |
| Slope | $-k$ | $-k$ | $+k$ |
| Half-Life ($t_{1/2}$) | $\frac{[A]_0}{2k}$ | $\frac{0.693}{k}$ | $\frac{1}{k[A]_0}$ |
Key Graphing Rules:
- If [A] vs t is straight, it is Zero Order.
- If ln[A] vs t is straight, it is First Order.
- If 1/[A] vs t is straight, it is Second Order.
Half-Life ($t_{1/2}$)
Time required for concentration to decrease to half its initial value.
- First Order Half-Life: This is the most important for AP Chem. It is constant. It does not depend on concentration. Radioactive decay is always 1st order.
- Zero/Second Order Half-Life: These depend on initial concentration. As the reaction proceeds and $[A]$ drops, $t_{1/2}$ changes (gets shorter for zero order, longer for second order).
5.5 Collision Theory
For a reaction to occur, particles must collide. However, not every collision results in a reaction. A successful "effective" collision requires two things:
- Correct Orientation: Particles must hit each other at the right a specific angle (active sites must align).
- Sufficient Energy ($Ea$): Particles must collide with kinetic energy $\ge$ Activation Energy ($Ea$).
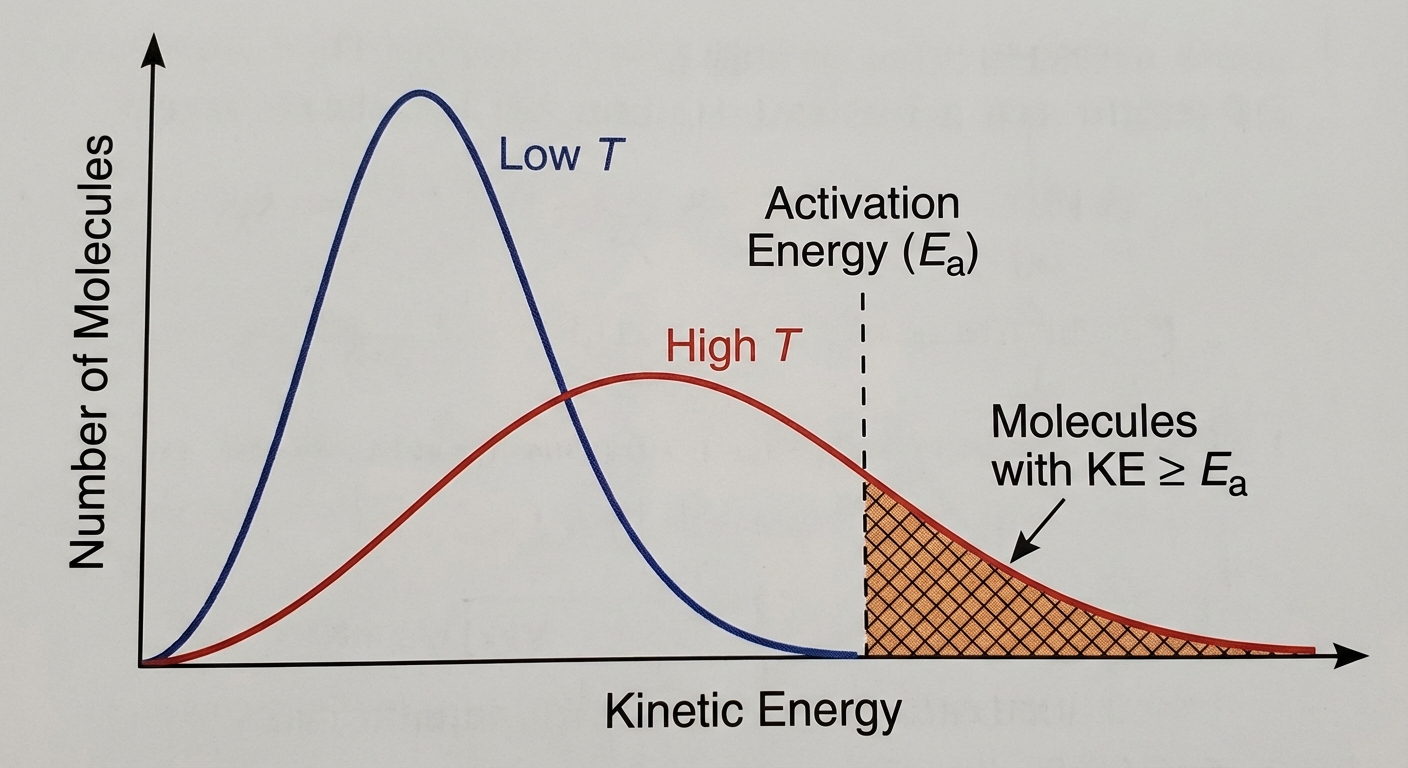
The Maxwell-Boltzmann Distribution
Temperature is a measure of average kinetic energy. At any temperature, molecules have a range of speeds.
- As Temperature increases, the distribution curve flattens and shifts to the right.
- The area under the curve representing particles with Energy $> E_a$ increases significantly.
- This explains why a small increase in $T$ leads to a large increase in Rate.
The Arrhenius Equation
Mathematical relationship between Rate Constant ($k$) and Temperature ($T$):
- $A$: Frequency factor (frequency of collisions + orientation probability).
- $E_a$: Activation Energy (Joules).
- $R$: Gas constant ($8.314 \text{ J}/\text{mol}\cdot\text{K}$).
- $T$: Temperature (Kelvin).
Takeaway: As $T$ increases, the exponent becomes less negative, so $k$ increases exponentially.
5.7 - 5.9 Reaction Mechanisms
Most reactions do not occur in a single step. They occur via a series of Elementary Steps called a mechanism.
Key Terms
- Elementary Step: Describes a single molecular event.
- Molecularity: Number of molecules colliding in an elementary step.
- Unimolecular: $A \rightarrow P$ (Rate $= k[A]$).
- Bimolecular: $A+B \rightarrow P$ (Rate $= k[A][B]$) or $A+A \rightarrow P$ (Rate $= k[A]^2$).
- Termolecular: 3 molecules colliding (Very rare).
- Intermediate: Produced in one step, consumed in a later step. (Does not appear in overall balanced equation or rate law).
- Catalyst: Present at the start, consumed, then produced again at the end. (Appears in rate law, not in balanced equation).
The Rate-Determining Step (RDS)
The slowest step in the mechanism determines the overall reaction rate.
Rule: The rate law of the overall reaction is equal to the rate law of the slow step (provided it doesn't contain intermediates).
Case 1: Slow Step is First
Step 1: $NO2 + NO2 \rightarrow NO3 + NO$ (Slow) Step 2: $NO3 + CO \rightarrow NO2 + CO2$ (Fast)
- Rate Law: Rate $= k1[NO2]^2$
- Since the slow step involves only reactants, we are done. Matches experiment.
Case 2: Fast Equilibrium Step (Harder)
Sometimes the slow step follows a fast reversible equilibrium. You cannot leave an intermediate in the final rate law.
Mechanism:
- $NO + Cl2 \rightleftharpoons NOCl2$ (Fast Equilibrium)
- $NOCl_2 + NO \rightarrow 2NOCl$ (Slow)
Derivation:
- Write rate for slow step: Rate $= k2[NOCl2][NO]$.
- Problem: $[NOCl_2]$ is an intermediate.
- Use Step 1 Equilibrium: Rate${fwd}$ = Rate${rev}$.
- $k1[NO][Cl2] = k{-1}[NOCl2]$
- Solve for $[NOCl_2]$:
- $[NOCl2] = \frac{k1}{k{-1}}[NO][Cl2]$
- Substitute back into slow step rate:
- Rate $= k2 \left( \frac{k1}{k{-1}}[NO][Cl2] \right) [NO]$
- Rate $= k{obs}[NO]^2[Cl2]$
5.6 & 5.11 Energy Profiles & Catalysis
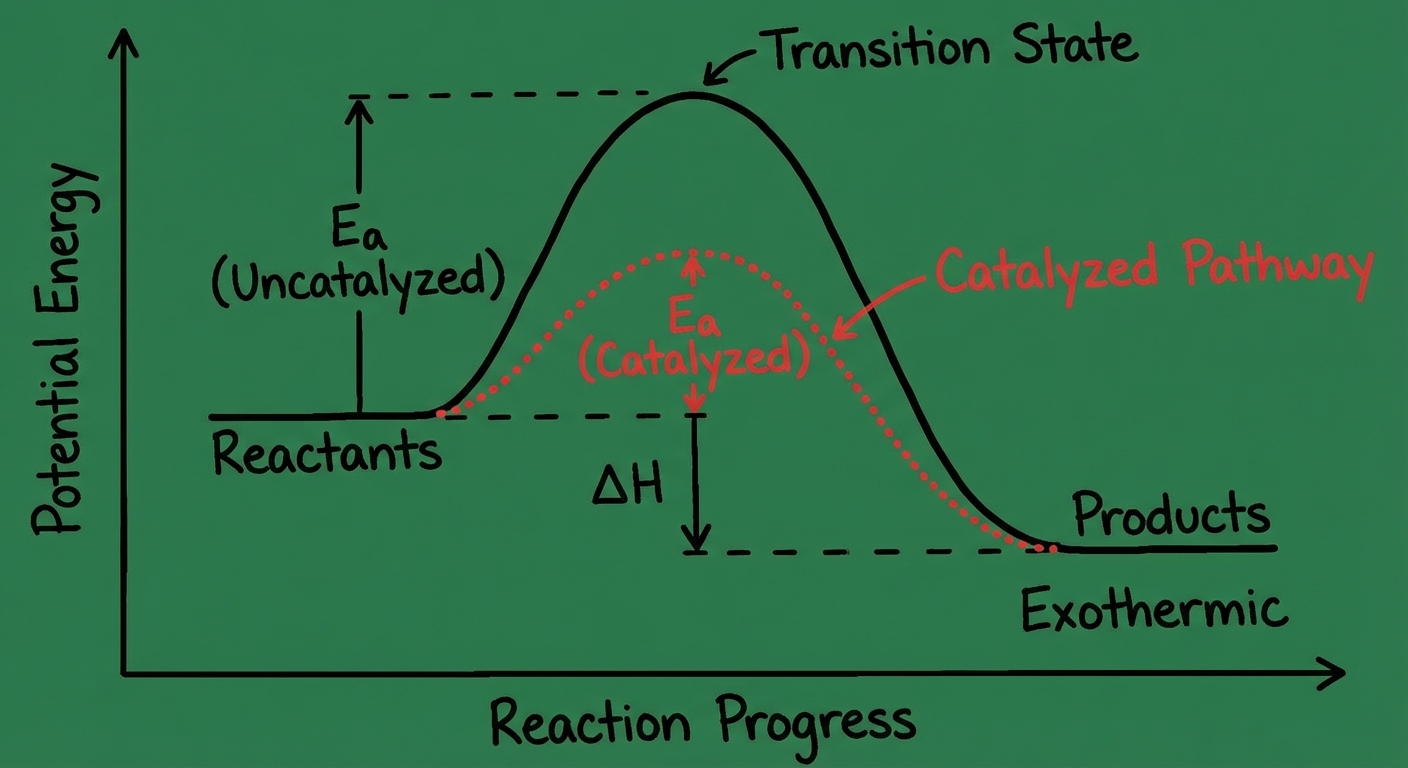
Reaction Energy Profiles
Graphs showing Energy vs. Reaction Progress.
- Activation Energy ($E_a$): The hill you must climb. Energy difference between Reactants and Transition State (activated complex).
- Enthalpy Change ($\Delta H$): Energy difference between Reactants and Products.
- Products lower than Reactants = Exothermic.
- Products higher than Reactants = Endothermic.
Multistep Profiles
A reaction with 2 elementary steps will have 2 "humps" (activation energy barriers) and a valley in between.
- The valley represents the Intermediate.
- The Highest Peak represents the activation energy of the Rate Determining Step.
Catalysts
Catalysts speed up reactions by lowering the Activation Energy ($E_a$).
- They creates a new reaction pathway (mechanism) with a lower energy barrier.
- Important: Catalysts do NOT change $\Delta H$ (thermodynamics) or the Equilibrium Constant ($K$). They only change the rate ($k$).
Types of Catalysts
- Acid-Base Catalysis: Reactant gains/loses a proton to become more reactive.
- Surface Catalysis: Reactants adsorb onto a metal surface (increasing collision probability), react, then desorb.
- Enzymes: Biological catalysts binding substrates at active sites.
Common Mistakes & Mnemonics
Mnemonics
- "L-N over 1 over": Helps remember the y-axes for 0, 1, 2 order graphs.
- 0 order: $[A]$ (Letters)
- 1st order: $\ln[A]$ (Natural Log)
- 2nd order: $1/[A]$ (One over)
Common Pitfalls
- Confusing Rate with Rate Constant:
- Mistake: "The catalyst increased $k$, so the rate increased." (Correct).
- Mistake: "Increasing concentration increases $k$." (WRONG. Only Temperature and Catalysts change $k$. Concentration changes Rate, not $k$).
- Stoichiometric Coefficients as Orders:
- Mistake: Seeing $2A + B \rightarrow C$ and assuming Rate $= k[A]^2[B]$.
- Correction: Orders must be determined experimentally. This assumption plays true ONLY for elementary steps, not overall reactions.
- Intermediate vs. Catalyst:
- Mistake: Mixing them up.
- Check: Catalyst: Start $\rightarrow$ End. Intermediate: Middle $\rightarrow$ Gone.
- Integrated Law Sign Errors:
- Remember that the slope for 0 and 1st order is negative ($-k$), but for 2nd order is positive ($k$).
- Wrong Units for $k$:
- Always check that (Units of Concentration)$^{\text{order}} \times$ (Units of $k$) = $M/s$.