AP Chemistry Unit 9: Entropy, Free Energy, and Favorability
Introduction to Entropy
Thermodynamics is the study of energy and its interconversions. While Enthalpy ($\Delta H$) tells us about heat flow, it isn't enough to predict if a reaction will happen naturally. To do that, we need a new quantity.
Defining Entropy ($S$)
Entropy ($S$) is a measure of the dispersal of matter and energy. Often simplified as "disorder" or "randomness," defined more rigorously in AP Chemistry involves microstates.
- Microstates: The specific configuration of all atom positions and energies at a given instant. High entropy means a system has a vast number of possible microstates.
- Second Law of Thermodynamics: The entropy of the universe increases in any thermodynamically favorable (spontaneous) process. \Delta S{univ} = \Delta S{sys} + \Delta S_{surr} > 0
States of Matter and Molecular Complexity
Entropy depends heavily on the physical state and structure of the substance.
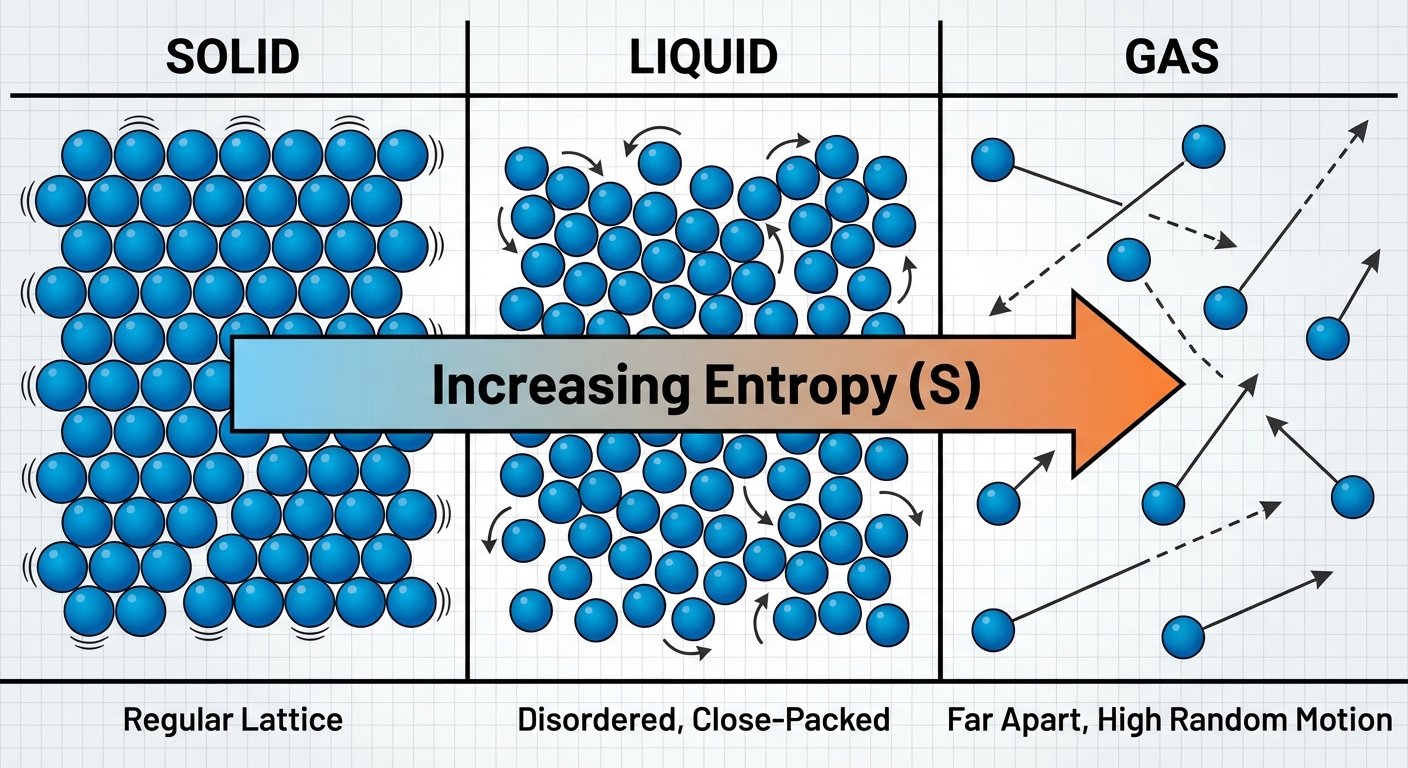
| Factor | Effect on Entropy ($S$) | Reasoning |
|---|---|---|
| State of Matter | $S{gas} \gg S{liquid} > S_{solid}$ | Gases have much more freedom of motion and spatial dispersal. |
| Temperature | Increases as $T$ increases | Higher $T$ means higher kinetic energy and more distribution of molecular speeds. |
| Volume (Gases) | Increases as $V$ increases | More space means more possible positions for particles (more microstates). |
| Complexity | Increases with size/molar mass | More electrons and protons, and more vibrational modes in bonds. |
Absolute Entropy and Entropy Change
Unlike Enthalpy ($H$), absolute Entropy ($S$) can be measured.
The Third Law of Thermodynamics
The entropy of a perfect crystal at absolute zero (0 K) is exactly zero. This provides a baseline reference point. Note: Elements in their standard states have non-zero standard entropies (unlike $\Delta H_f^\circ$, which is zero for pure elements).
Calculating $\Delta S^\circ_{rxn}$
To calculate the standard entropy change for a reaction, we use Hess's Law logic:
- Positive $\Delta S$ ($+S$): Matter is becoming more dispersed (e.g., solid $\rightarrow$ gas, or $2$ gas moles $\rightarrow$ $4$ gas moles).
- Negative $\Delta S$ ($-S$): Matter is becoming more ordered/constrained (e.g., gas $\rightarrow$ liquid, precipitation).
Gibbs Free Energy and Thermodynamic Favorability
Gibbs Free Energy ($G$) combines enthalpy and entropy to predict thermodynamic favorability (formerly called spontaneity). Favorability means a process will occur without continuous external intervention.
The Gibbs-Helmholtz Equation
This is arguably the most critical equation in Unit 9:
- Units: Be careful! $\Delta H$ is usually in kJ/mol, while $\Delta S$ is in J/mol$\,\cdot\,$K. You must convert $\Delta S$ to kJ or $\Delta H$ to J before subtracting.
- Standard Conditions: $T = 298\,K$, $1\,atm$, $1\,M$.
Interpreting Signs
The sign of $\Delta G$ determines favorability:
- $\Delta G < 0$: Thermodynamically favorable (spontaneous).
- $\Delta G > 0$: Thermodynamically unfavorable (non-spontaneous).
- $\Delta G = 0$: The system is at equilibrium.
Temperature Dependence of $\Delta G$
The signs of $\Delta H$ and $\Delta S$ determine at which temperatures a reaction is favorable.
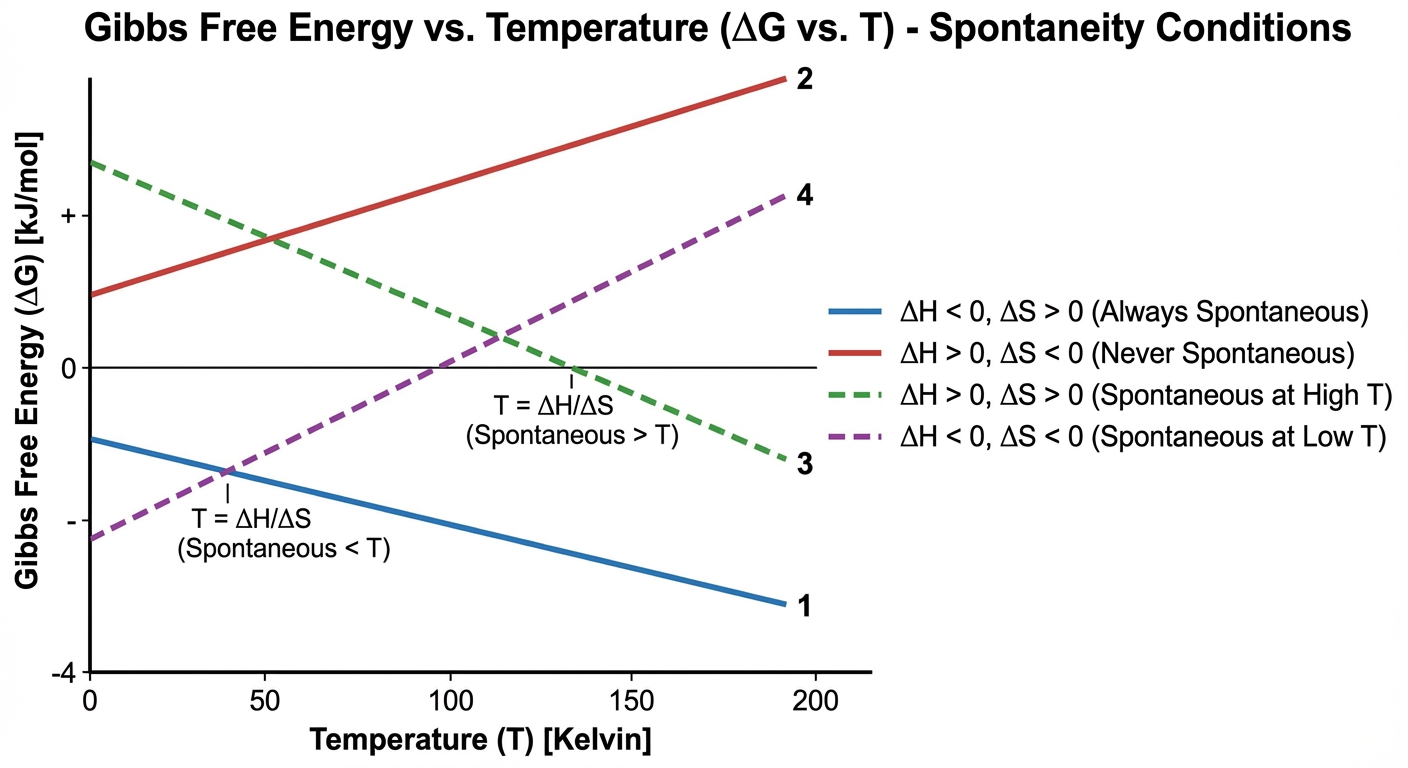
| $\Delta H$ | $\Delta S$ | Outcome for $\Delta G$ | Favorability |
|---|---|---|---|
| $-$ (Exothermic) | $+$ (Dispersing) | Always Negative | Favorable at ALL temperatures |
| $+$ (Endothermic) | $-$ (Ordering) | Always Positive | Favorable at NO temperatures |
| $-$ (Exothermic) | $-$ (Ordering) | Negative only at Low $T$ | Favorable at LOW temperatures |
| $+$ (Endothermic) | $+$ (Dispersing) | Negative only at High $T$ | Favorable at HIGH temperatures |
Mnemonic: To remember High/Low cases, look at $\Delta S$. If $\Delta S$ is positive, you want high $T$ to make the $-T\Delta S$ term large enough to dominate.
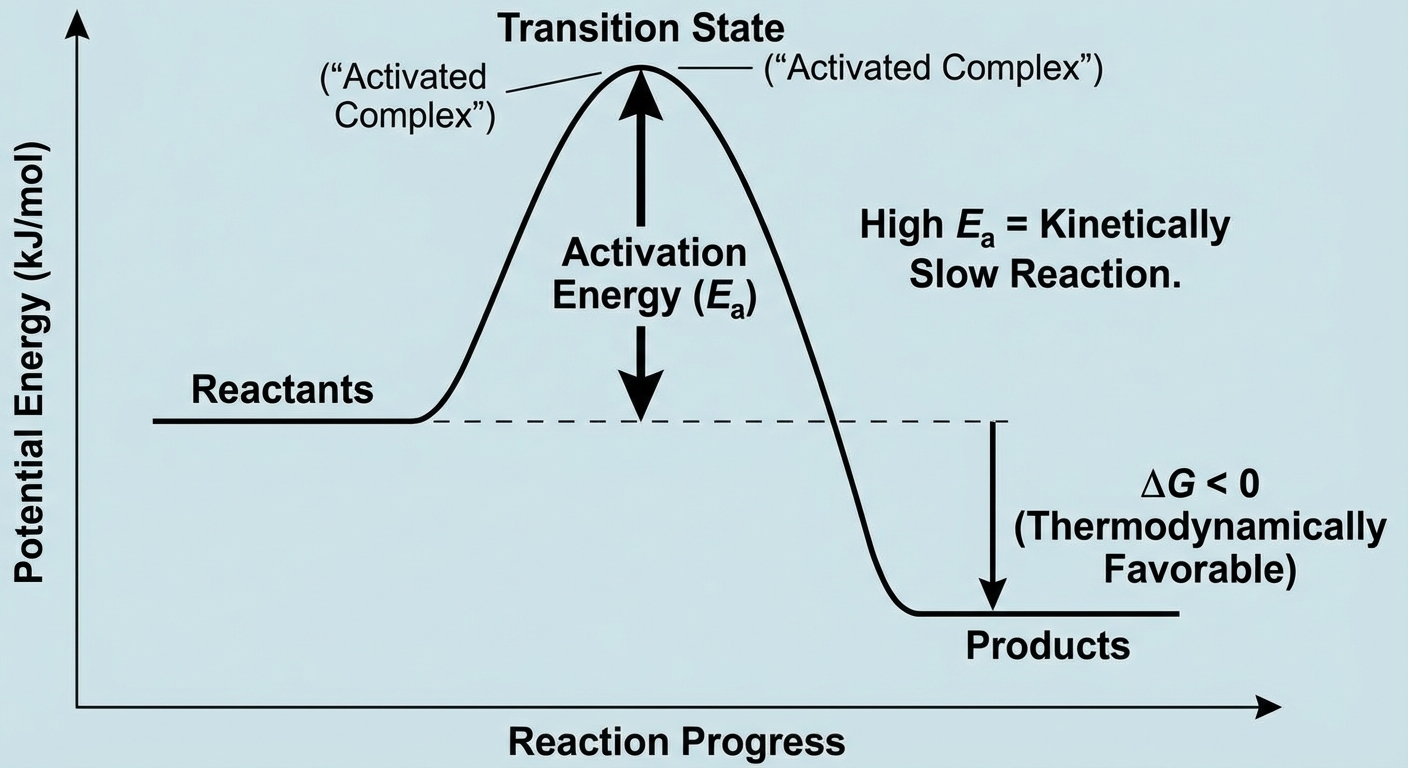
Thermodynamic vs. Kinetic Control
Just because a reaction is "thermodynamically favorable" ($\Delta G < 0$) does not mean it will happen quickly.
Kinetic Control
A reaction is under kinetic control if it has a large negative $\Delta G$ (should happen) but occurs at a negligible rate.
- Reason: High Activation Energy ($E_a$).
- Example: Diamond turning into graphite is thermodynamically favorable ($\Delta G < 0$), but the reaction is so slow (millions of years) that diamonds are effectively stable. We say this reaction is kinetically controlled.
Free Energy and Equilibrium
$\Delta G^\circ$ tells us about the favorability from standard states to equilibrium. The Equilibrium Constant ($K$) tells us how far the reaction proceeds.
The Relationship
The bridge between Thermodynamics and Equilibrium is calculated as:
Where:
- $R = 8.314\, J/(mol\cdot K)$ (Gas constant in energy units)
- $T =$ Temperature in Kelvin
- $K =$ Equilibrium constant ($K{eq}$, $Kp$, $K_c$, etc.)
Qualitative Implications
- If $\Delta G^\circ < 0$, then $\ln K > 0$, so $K > 1$. The equilibrium favors products.
- If $\Delta G^\circ > 0$, then $\ln K < 0$, so $K < 1$. The equilibrium favors reactants.
- If $\Delta G^\circ = 0$, then $\ln K = 0$, so $K = 1$.
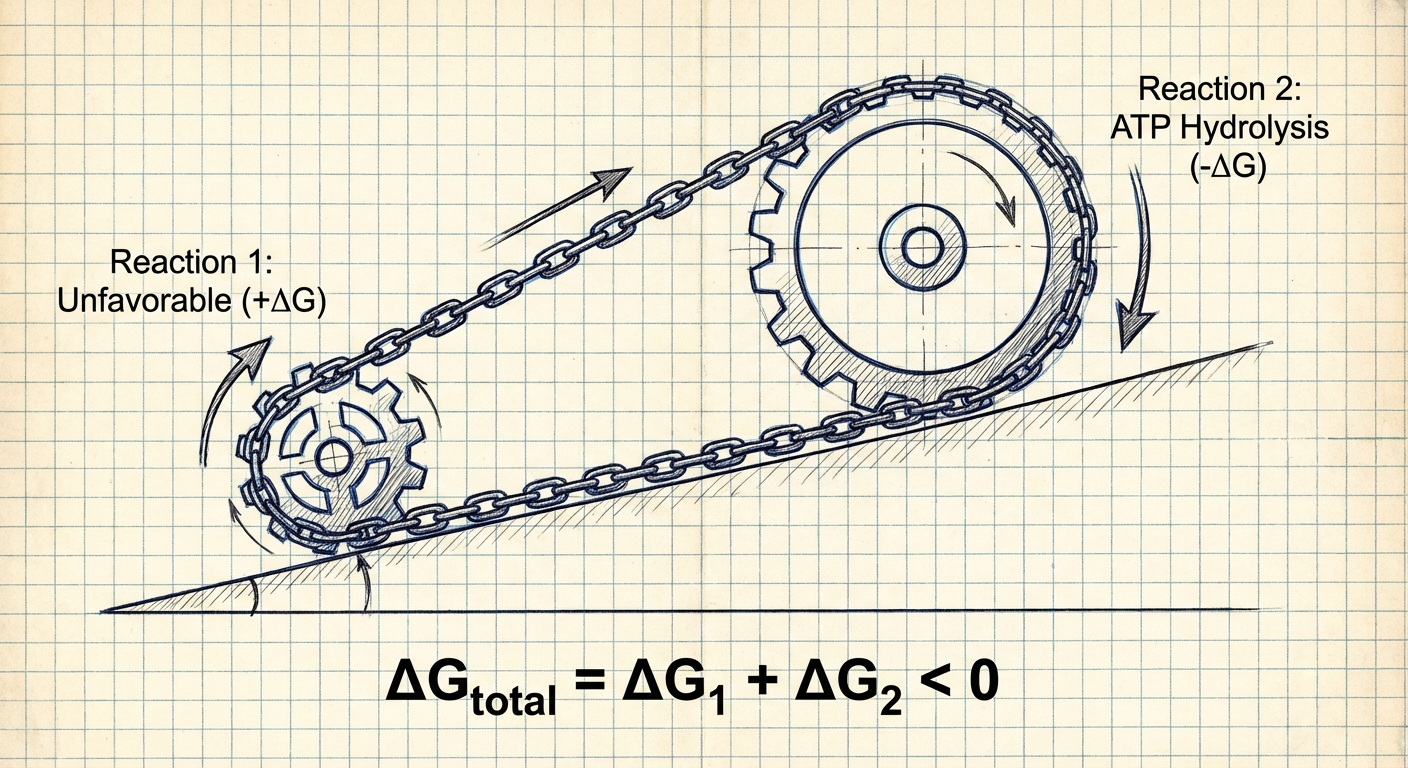
Coupled Reactions
Biological systems and industrial processes often need to perform reactions that are thermodynamically unfavorable ($\Delta G > 0$). To achieve this, they use coupled reactions.
The Principle
Two reactions that share a common intermediate can be coupled. If you add the reactions together, you add their $\Delta G$ values (Hess's Law).
- Reaction A (Unfavorable): $\Delta G_1 = +20\,kJ$
- Reaction B (Highly Favorable): $\Delta G_2 = -50\,kJ$
Net Reaction: $\Delta G_{total} = +20 + (-50) = -30\,kJ$
Because the net $\Delta G$ is negative, the overall process becomes favorable.
- Example: ATP hydrolysis ($
\Delta G < 0$) is often coupled with protein synthesis ($ \Delta G > 0$) in the body to drive the synthesis forward.
Common Mistakes and Pitfalls
- The Unit Trap: Students consistently fail to convert units in $\Delta G = \Delta H - T\Delta S$.
- $\Delta H$ is usually kJ.
- $\Delta S$ is usually J.
- Fix: Divide $\Delta S$ by 1000 to get kJ/K before calculating.
- "Spontaneous" Confusion: Don't assume "spontaneous" means "fast." Rusting is spontaneous, but slow. Always distinguish between Thermodynamics (Direction) and Kinetics (Rate).
- Standard Conditions vs. Equilibrium: $\Delta G^\circ$ is the free energy change at standard conditions (all pure, 1M, 1atm). At equilibrium, calculate $\Delta G$ (without the degree symbol), which equals 0. Do not confuse $\Delta G^\circ$ with $\Delta G$.
- Temperature Units: Using Celsius instead of Kelvin. Always use $K = ^\circ C + 273.15$.